В случае взаимодействия двух атомов:
U – энергия взаимодействия;
U = U ПРИТЯЖ. + U ОТТАЛК.
 - уравнение
Леннарда-Джонса
,
c,
b,
m
= const
- уравнение
Леннарда-Джонса
,
c,
b,
m
= const
В случаях взаимодействия атомов с твердой поверхностью, необходимо провести суммирование всех взаимодействий.
х– расстояние до поверхности
r – радиус действия сил притяжения
dV – объем
n – число молекул поверхности
U АДС. – энергия адсорбционного взаимодействия



В случае адсорбции притяжение усиливается. И в случае при взаимодействии типа неполярное-неполярное адсорбция преимущественно локализуется в углублениях.
Электростатическое взаимодействие.
Полярный адсорбент – неполярный адсорбат
Неполярный адсорбент – полярный адсорбат
Полярный адсорбент – полярный адсорбат.
М олекулу
адсорбата представляют как диполь, а
адсорбента – как проводник, в котором
молекула адсорбата индуцирует диполь
зеркально симметрично по отношению к
данному.
олекулу
адсорбата представляют как диполь, а
адсорбента – как проводник, в котором
молекула адсорбата индуцирует диполь
зеркально симметрично по отношению к
данному.
X – расстояние до середины
При взаимодействии возникает потенциал:
 ,
,
 - дипольный момент.
- дипольный момент.
Потенциал стремится принять максимальное значение, т.е. диполи стремятся сориентироваться перпендикулярно к поверхности.
Поскольку повышение температуры способствует росту Броуновского движения, оно приводит к торможению процесса адсорбции.
В случае электростатического взаимодействия адсорбат преимущественно локализуется на выступах.
Фундаментальное адсорбционное уравнение.
В случае адсорбции происходит перераспределение компонента, а значит, изменяется химический потенциал. Процесс адсорбции можно рассматривать как переход поверхностной энергии в химическую.
Объем слоя = 0, тогда обобщенное уравнение I и II закона термодинамики:
T
= const;
(1) = (2) =>


Для двухкомпонентной системы:
,
 ,
,
 =>
=>
=>
 - адсорбционное
уравнение Гиббса
.
- адсорбционное
уравнение Гиббса
.
Для случая адсорбции
тв. тело – газ:
,
 ,
,

 - изотерма
- изотерма
 - изобара
- изобара
 - изопикна
- изопикна
 - изостера
- изостера
Изотерма, изопикна, изостера связаны друг с другом.
Т.к. адсорбция
функция




Изотерма Генри Изотерма Лангмюра



Термодинамика. Адсорбция.
Для конденсированных сред:
 ,
,
 ,
,
 - интегральное
изменение энергии Гиббса
.
- интегральное
изменение энергии Гиббса
.

P–давление над искривленной поверхностью, Р S –давление над плоской поверхностью
 - адсорбционный
потенциал
- адсорбционный
потенциал
Дифференциальное
изменение энтрапии

 ,
Г = const
,
Г = const
- дифференциальное изменение энтропии
- дифференциальная энтальпия адсорбции
 - изостерическая
теплота адсорбции
- изостерическая
теплота адсорбции
 - теплота
конденсации
- теплота
конденсации
 - чистая
теплота адсорбции
- чистая
теплота адсорбции

 ,
,




Qa
– интегральная теплота адсорбции,

Qra – интегральная чистая теплота адсорбции,

Уравнение Генри
Исследование адсорбции затрудняется неоднородностью поверхности, поэтому простейшие закономерности получают для однородных поверхностей.
Рассмотрим взаимодействие газов с твердой поверхностью, когда осуществляется переход газа из равновесного состояния в объеме в равновесное состояние на поверхности. Этот случай аналогичен равновесию газов в поле силы тяжести.
 ,
,
 ,
=>
,
=> -уравнение
Генри
-уравнение
Генри
 - коэффициент
распределения
- коэффициент
распределения
В процессе адсорбции происходит изменение химических потенциалов.
Для объемной фазы:

Для газа на
поверхности:

В состоянии
равновесия
 ,
т.е.
,
т.е.

В уравнении Генри константа не зависит от концентрации
Уравнение Генри выполняется в области низких давлений и концентраций. По мере роста концентрации возможны 2 типа отклонений от закона Генри:
1 – положительные отклонения, D уменьшается, А уменьшается
2 – отрицательные отклонения, D – возрастает, А – возрастает.
Тип отклонения определяется преобладанием тот или иного типа взаимодействия адсорбент-адсорбат.
При сильном адгезионном взаимодействии коэффициенты активности возрастают – положительное отклонение. В случае когезионных взаимодействий наблюдаются отрицательные отклонения.
Мономолекулрная адсорбция.
Изотерма Лангмюра.
Простейшие закономерности были получены в теории генри. Ленгмюр предложил теорию, согласно которой, адсорбция рассматривается как квазихимическая реакция. При этом:
Поверхность энергетически однородна.
Адсорбция локализована, каждый адсорбционный центр взаимодействует с одной молекулой адсорбата.
Молекулы адсорбата не взаимодействуют друг с другом.
Адсорбция монослойна.

 - поверхность,
- поверхность, - адсорбат,
- адсорбат, - адсорбционный комплекс.
- адсорбционный комплекс.
 ,
тогда концентрация адсорбционных мест:
,
тогда концентрация адсорбционных мест:
 ,
, - предельная адсорбция.
- предельная адсорбция.
 ,
тогда константа реакции:
,
тогда константа реакции:


 - уравнение Лангмюра.
- уравнение Лангмюра.
Зависимость адсорбции от концентрации
1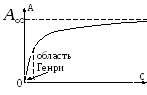 )
)
 ,
,

2) область высоких концентраций
 - предельная
адсорбция, образование мономолекулярного
слоя
- предельная
адсорбция, образование мономолекулярного
слоя
Для энергии Гиббса: .
g – энтропийный множитель.
В случае изотермы
Генри энергия Гиббса характеризует
переход адсорбата из стандартного
состояния в объёме в стандартное
состояние на поверхности. В случае
изотермы Ленгмюра
 характеризует степень сродства адсорбента
и адсорбата.
характеризует степень сродства адсорбента
и адсорбата.
 находят из изобары
Вант-Гоффа.
находят из изобары
Вант-Гоффа.

 ,
тогда
,
тогда
 ,
отсюда
,
отсюда .
.

 - степень заполнения
поверхности.
- степень заполнения
поверхности.




 - число свободных
мест,
- число свободных
мест,
 - число занятых мест.
- число занятых мест.
 ,
,

Т.е. в области высоких концентраций число свободных мест обратно пропорционально количеству адсорбата.
Адсорбция смеси газов на однородной поверхности.
В этом случае процесс адсорбции рассматривают как две параллельно протекающие реакции.
 (1)
(1)

 (2)
(2)


Адсорбция смеси газов на неоднородной поверхности.
В случае неоднородной поверхности нельзя ограничиваться средними заполнениями.
В следствие конкурентной борьбы, на участках различных типов возможна локализация различных адсорбатов.
В этом случае
отношение
 .
.
 ,
,
 - давление насыщенного пара адсорбата.
- давление насыщенного пара адсорбата.
 ,
,
 - теплоты адсорбции.
- теплоты адсорбции.
«+» - симбатная зависимость, «-» - антибатная зависимость, «Н» - корреляции нет.
«+» - адсорбция протекает по одинаковому механизму. На наиболее энергетически выгодных участках преимущественно адсорбируется газ, обладающий большим сродством к поверхности.
«-» - адсорбция протекает по различным механизмам и до определенного момента времени конкурентной борьбы за поверхность нет.
Мономолекулярная адсорбция преимущественно реализуется при физической адсорбции газов при малых значениях p , а также на границе раздела жидкость/газ.
Полимолекулярная адсорбция.
Теория БЭТ (Брунауэр, Эммет, Теллер).
В случае, когда образование монослоя недостаточно для компенсации поверхностной энергии, адсорбция полимолекулярна и её можно рассматривать как результат вынужденной конденсации под действием поверхностных сил.
Основные положения:
При попадании молекулы адсорбата на занятое место образуется кратный комплект.
По мере приближения p к p s уменьшается число свободных адсорбционных мест. Первоначально увеличивается, а затем уменьшается число мест, занятых единичными, двойными и т.д. комплектами.
При p =p s адсорбция переходит в конденсацию.
Горизонтальные взаимодействия отсутствуют.
Для первого слоя выполняется изотерма Лангмюра.
Поверхность рассматривается как совокупность адсорбционных мест. Справедливо условие динамического равновесия: скорость конденсации на свободных местах равна скорости испарения с занятых.
a – коэффициент конденсации (доля молекул, сконденсировавшихся на поверхности);
 ,
,
Zm – максимальное число свободных мест.
 -
частота колебаний атомов в направлении
перпендикулярном к поверхности.
-
частота колебаний атомов в направлении
перпендикулярном к поверхности.
Для первого слоя условия динамического равновесия:



 ,
тогда
,
тогда
 - уравнение Лангмюра.
- уравнение Лангмюра.
Для второго слоя
будет справедливо:

Для i-го
слоя:

Для упрощения принимают, что а и ν одинаковы для всех слоев, кроме первого. Для всех слоев кроме первого теплота адсорбции постоянна. Для последнего слоя теплота адсорбции равна теплоте конденсации. В результате было получено уравнении
 (*)
(*)
C – константа,
В случае теории БЭТ, константа С характеризует энергию Гиббса чистой адсорбции. Уравнение содержит только одну константу, а также это уравнение очень важно для определения удельной поверхности адсорбента.
Поскольку в результате адсорбции теплота выделяется, определение удельных поверхностей ведут при низких температурах.
 ????????????
????????????


Основной недостаток теории – пренебрежение горизонтальными взаимодействиями в пользу вертикальных.
Уравнение выполняется
в области значений
 от 0,05 до 0,3.
от 0,05 до 0,3.
Там, где
 <
0,05 – существенное влияние оказывает
неоднородность поверхности.
<
0,05 – существенное влияние оказывает
неоднородность поверхности.
 >
0,3 – сказывается взаимодействие адсорбат
– адсорбат.
>
0,3 – сказывается взаимодействие адсорбат
– адсорбат.
Учет взаимодействий адсорбат-адсорбат.
Взаимодействия проявляются при адсорбции на неполярной поверхности разветвленных молекул или молекул. Способных образовывать ассоциаты. В этом случае изменяется форма изотерм адсорбции.
А дсорбент
не полярен.
дсорбент
не полярен.
Графику 1 соответствуют слабые взаимодействия адсорбат-адсорбат, сильное адсорбат-адсорбент.
Графику 2 соответствуют сильное взаимодействие адсорбат-адсорбат, сильное адсорбат-адсорбент.
Графику 3 соответствуют сильное взаимодействие адсорбат-адсорбат, слабое адсорбат-адсорбент.
 ,
,

В случае взаимодействия между молекулами адсорбата необходим учет изменения коэффициентов активности. И это уравнение записывают в виде:
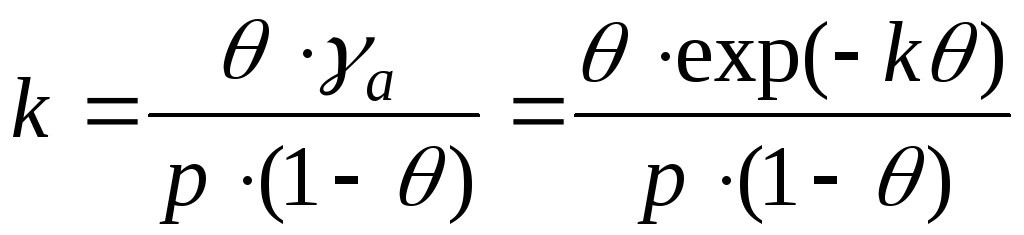 - уравнение Фрункина,
Фаулера, Гугенгейма.
- уравнение Фрункина,
Фаулера, Гугенгейма.
k – аттракционная постоянная.
Потенциальная теория Поляни.
Данная теория не выводит какого-либо типа изотермы адсорбции, а дает возможность расчета изотерм при другой температуре.
Адсорбция – это результат притяжения адсорбата к поверхности адсорбента за счет действия адсорбционного потенциала, который не зависит от присутствия других молекул и зависит от расстояния между поверхностью и молекулой адсорбата.
 ,
,
 - адсорбционный потенциал.
- адсорбционный потенциал.
Поскольку поверхность
неоднородная, расстояние заменяют на
адсорбционный объём
 .Адсорбционный
объём
– это
объём, заключенный между поверхностью
и точкой, соответствующей данному
значению
.Адсорбционный
объём
– это
объём, заключенный между поверхностью
и точкой, соответствующей данному
значению .
.
Адсорбционный потенциал – это работа перенесения 1 моль адсорбата вне данного адсорбционного объёма в данную точку адсорбционного объёма (или работа переноса 1 моль насыщенного пара адсорбата, находящегося в равновесии с жидким адсорбатом в отсутствии адсорбента в равновесную с адсорбентом паровую фазу).

Характеристическая кривая
 - адсорбционный
потенциал,
- адсорбционный
потенциал,

Для данного
адсорбента и различных адсорбатов
справедливо:

Для разных типов
адсорбатов
 ,
,
где
 потенциалы для изотерм адсорбции при
относительных давлениях
потенциалы для изотерм адсорбции при
относительных давлениях для
адсорбата 1 и для адсорбата 2. Это отношение
есть величина постоянная.
для
адсорбата 1 и для адсорбата 2. Это отношение
есть величина постоянная.
 - коэффициент
аффинности
- коэффициент
аффинности
Теория капиллярной конденсации.
Протекание процесса адсорбции во многом зависит от структуры пористого тела.
|
Микропористые | |
|
Переходнопористые | |
|
Макропористые |
В случае микропористых
сорбентов, поля адсорбционных сил
перекрываются. В случае макропористых
сорбентов, поры выполняют роль транспортных
каналов. Процессы коденсации наиболее
значимы в переходнопористых телах.
Капиллярная конденсация начинается
при определенных значениях p
и
 ,
когда часть поверхностной энергии уже
скомпенсирована. Необходимое условие
– поверхность должна быть самчиваема.
Процее описываетсяуравнением
Томпсона – Кельвина
.
,
когда часть поверхностной энергии уже
скомпенсирована. Необходимое условие
– поверхность должна быть самчиваема.
Процее описываетсяуравнением
Томпсона – Кельвина
.

 - для случая
смачивания, центр кривизны находится
в газовой фазе.
- для случая
смачивания, центр кривизны находится
в газовой фазе.
В случае капиллярной конденсации изотерма адсорбции имеет гистерезисный вид. Процессу адсорбции соответствует нижняя ветвь, процессу десорбции – верхняя.
Все виды пор можно свести к трем видам:
|
Конические |
Цилиндрические с одним закрытым концом |
Цилиндрические с двумя открытыми концами |
|
Процессное заполнение осуществляется со дна поры. Изтерма адсорбции и изотерма десорбции в этом случае совпадают, поскольку процесс адсорбции начинается со сферы и процесс десорбции также начинается с исчезновения некоторых сфер.
↓ |
Гистерезиса нет. Прямой и обратный ход описываются уравнением:
|
Дна нигде нет, заполнение поры пойдет по стенкам цилиндра.
цилиндр: Изотерма и будет иметь гистерезисный вид.
↓ |


 - сфера,
- сфера, ,
,



В условиях смачивания конденсация
осуществляется при более низких
давлениях, что энергетически выгодно.
По десорбционной ветви получают кривые
распределения пор по размерам.
условиях смачивания конденсация
осуществляется при более низких
давлениях, что энергетически выгодно.
По десорбционной ветви получают кривые
распределения пор по размерам.
Максимум
дифференциальной кривой смещен влево
относительно точки перегиба интегральной.
Общий объём малых пор невелик, однако
имеет большие значения площади
поверхности. С увеличением размера пор,
их объём возрастает как
 ,
а площадь как
,
а площадь как ,
за счет этого и наблюдается смещение
максимума дифференциальной кривой.
,
за счет этого и наблюдается смещение
максимума дифференциальной кривой.
Адсорбция на границе твердое тело – жидкость.
В случае адсорбции на границе твердое тело – газ, мы пренебрегали одним компонентом. В случае адсорбции на границе твердое тело – жидкость адсорбат вытесняет с поверхности адсорбента молекулы растворителя.
 ,
,
Справедливо уравнение:

 ,
,
N 1 , N 2 – мольные доли растворителя и компонента, N 1 + N 2 = 1, тогда
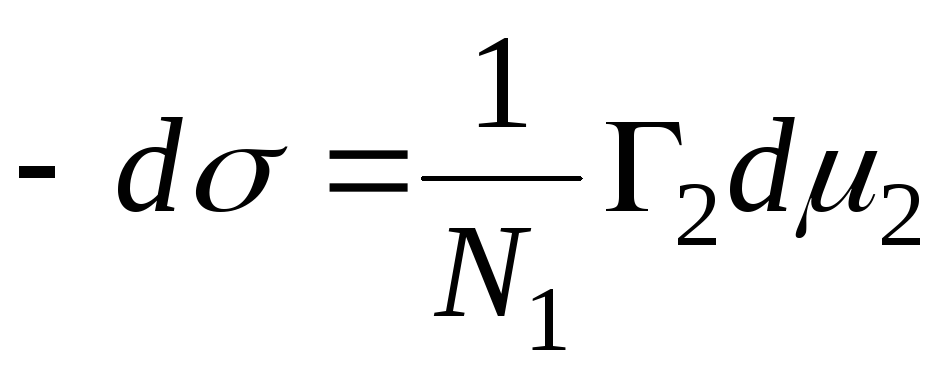 ,
=>
,
=>
 ,
тогда- уравнение адсорбции для границы раздела
фаз твердое тело – жидкость.
,
тогда- уравнение адсорбции для границы раздела
фаз твердое тело – жидкость.
Адсорбция (Г) >
0 при
 <
0
<
0
Если
значения для компонента и растворителя сильно
различны, в этом случае зависимостьГ
от N
имеет экстремум при значении N
~ 0,5.
для компонента и растворителя сильно
различны, в этом случае зависимостьГ
от N
имеет экстремум при значении N
~ 0,5.
Е сли
сли имеют
близкие значения, в этом случае возможно
изменение знака адсорбции. ЗависимостьГ
от N
пересекает ось абсцисс
имеют
близкие значения, в этом случае возможно
изменение знака адсорбции. ЗависимостьГ
от N
пересекает ось абсцисс
Точка пересечения функции Г (N ) с осью абсцисс называется адсорбционным азеотропом . Это значит, что два компонента не могут быть разделены на данном адсорбенте.
Уравнение изотермы адсорбции с константой обмена.
При адсорбции на границе твердое тело – жидкость постоянно происходит перераспределение компонентов между поверхностью адсорбента и объемом раствора.
 -
компоненты
(- - относятся к поверхности)
-
компоненты
(- - относятся к поверхности)

 ,
,
 ,
, .
.
 ,
,


Адсорбция на границе жидкость-газ
Р ассмотрим
изменение концентрационного профиля
по мере пересечения границы раздела
жидкость-газ. Пусть компонент 2 летуч.
ассмотрим
изменение концентрационного профиля
по мере пересечения границы раздела
жидкость-газ. Пусть компонент 2 летуч.
Cs – концентрация в поверхностном слое.
Исходя из определения избыточной адсорбции

Если компонент не летуч, то величина адсорбции запишется следующим образом:
П ри
ри

В уравнении
 природа вещества описывается производной
природа вещества описывается производной .
.
Изотерма поверхностного натяжения может иметь вид 1 или 2:
1 – поверхностноинактивные вещества
2 – поверхностноактивные вещества
Поверхностной активностью g называется способность веществ снижать поверхностное натяжение в системе.

 - толщина
поверхностного слоя
- толщина
поверхностного слоя
C s – концентрация компонента в поверхностном слое
С – объемная концентрация
Для гомологического ряда существует правило:
 - правило Траубо
Дюкло
- правило Траубо
Дюкло
Для гомологического ряда изотерма адсорбции выглядит таким образом:
Вместо A пишем Г, так как адсорбция избыточная в поверхностном слое.
Изотерма поверхностного натяжения:

 - поверхностное
натяжение чистого растворителя.
- поверхностное
натяжение чистого растворителя.
 - фундаментальное
адсорбционное уравнение;
- фундаментальное
адсорбционное уравнение;
 -
уравнение Лангмюра.
-
уравнение Лангмюра.
Решим их совместно:

- уравнение Шишковского.
B – константа для гомологического ряда.
A
- при переходе от одного гомолога к
другому увеличивается в 3-3,5 раза

![]()
1 – область малых концентраций
![]()
2 – средняя концентрация
3 – мономолекулярный слой
Поверхностноактивные вещества представляют собой дифильные молекулы, т.е. включают полярную группу и неполярный углеводородный радикал.
o - полярная часть молекулы.
| - неполярная часть молекулы.
В полярном растворителе молекулы ПАВ ориентируются таким образом, что полярная часть молекулы обращена к растворителю, а неполярная выталкивается в газовую фазу.
В уравнении
Шишковского
 ,
она постоянна для гомологического ряда.
,
она постоянна для гомологического ряда.
Поверхностноактивное действие начинает проявляться с n >5. При концентрациях больших, чем концентрация мономолекулярного слоя, в растворах ПАВ происходит мицеллообразоваие.
Мицелла – называется агрегат молекул дифильных ПАВ, углеводородные радикалы которых образуют ядро, а полярные группы обращены в водную фазу.
Масса мицеллы – мицелляльная масса.
Ч исло
молекул – число агрегации.
исло
молекул – число агрегации.
Сферические мицеллы
В случае мицеллообразования в растворе устанавливается равновесие
ККМ – критическая концентрация мицеллообразования.
Поскольку мы считаем мицеллу отдельной фазой:
Для гомологического ряда существует эмпирическое уравнение:
a – энергия растворения функциональной группы.
b
 – инкремент адсорбционного потенциала,
работа адсорбции на одно метиленовое
звено.
– инкремент адсорбционного потенциала,
работа адсорбции на одно метиленовое
звено.
Наличие в мицеллах углеводородного ядра создает возможность для растворения в водных растворах ПАВ соединений, которые не растворимы в воде, это явление называется солюбилизацией (то, что растворяется – солюбилизат, ПАВ – солюбилизатор).
Грязь может быть совсем не полярна, может содержать как полярную, так и не полярную часть и будет ориентироваться как молекула ПАВ.
В любом случае при солюбилизации происходит увеличение мицеллярной массы и числа агрегации не только за счет включения солюбилизата, но и за счет увеличения числа молекул ПАВ, необходимых для поддержания равновесного состояния.
Солюбилизация тем эффективнее, чем меньше молекулярная масса солюбилизата.

 ~
72 мН\м.
~
72 мН\м.
 ~
33 мН\м.
~
33 мН\м.
Эффективность ПАВ зависит от величины ККМ.
Двухмерное давление поверхностного слоя

→ -силы поверхностного натяжения.
- двухмерное давление.
Поверхностный слой – это сила равная разности поверхностных натяжений раствора ПАВ и чистого растворителя, направленная в сторону чистой поверхности.
Между раствором и поверхностным слоем устанавливается равновесие



При
 существует область, где
существует область, где линейно зависит
от концентрации.
линейно зависит
от концентрации.
Г [моль/м 2 ].
 -площадь, занимаемая
одним молем вещества
-площадь, занимаемая
одним молем вещества
Тогда изотерма
двухмерного давления будет иметь вид

 - изотерма двухмерного
давления.
- изотерма двухмерного
давления.
Зависимость
 отS М:
отS М:

При
 - двухмерное давление резко возрастает.
При
- двухмерное давление резко возрастает.
При двухмерный деформируется, что вызывает
резкий рост
двухмерный деформируется, что вызывает
резкий рост .
.
Пленка с обеих сторон ограниченная одинаковыми фазами называется двусторонней. В таких пленках наблюдается постоянное движение маточного раствора.
Пленки толщиной меньше 5 нм называются черными пленками.

Адсорбционные слои должны обладать двумя характеристиками: вязкость и легкоподвижность, текучесть и упругость.
Эффект Марангони – это самозалечивание.
Треугольник Гиббса,
 - избыточное давление.
- избыточное давление.
Пленка растянулась и за счет того, что часть жидкости ушла, ПАВ усремляются в свободное место. Треугольник Гиббса.
Эффект адсорбционной прочности тел.
На поверхности пленки всегда существует адсорбционный слой, для которого , тогда
Уравнение Лангмюра:


 в двухмерное
давление
в двухмерное
давление
 - аналог уравнения
Шишковского
- аналог уравнения
Шишковского
Электрокинетические явления. Двойной электрический слой (ДЭС).
Модель Гелемгольца. Теория Гуи-Чапмена.
1808 г. Рейс

U – образная трубка, погружают в неё 2 электрода. Закон сообщающихся сосудов нарушается и происходит изменение уровня жидкости в трубке – электрокинетические явления.
Кинетические явления:
Электрофорез
Электроосмос
Потенциал течения (протекания)
Потенциал седиментации
1 и 2 возникают при наложении разности потенциалов, 3 и 4 продавливание и седиментация коллоидных частиц вызывают появление разности потенциалов.
Электроосмосом называется движение дисперсионной среды относительно неподвижной дисперсной фазы под действием электрического тока.
Электрофорез – это движение частиц дисперсной фазы относительно неподвижной дисперсионной среды под действием электрического тока.
П ричина
возникновения электрокинетических
явлений – это пространственного
разделение зарядов и возникновение
двойного электрического слоя.
ричина
возникновения электрокинетических
явлений – это пространственного
разделение зарядов и возникновение
двойного электрического слоя.
Двойной электрический
слой представляет собой плоский
конденсатор, одна обкладка образована
потенциалоределяющими ионами, другая
– противоиноами. Ионы заражены также
как потенциалопределяющие ко-ионы
оттеснены в объем раствора. Расстояние
между обкладками
 .
Потенциал падает линейно, разность
потенциалов
.
Потенциал падает линейно, разность
потенциалов .
.
Внешняя разность
потенциалов вызывает появление модуля
сдвига
 - это пара сил отнесенных к единице
площади, девствующих вдоль поверхности
твердого тела.
- это пара сил отнесенных к единице
площади, девствующих вдоль поверхности
твердого тела.

В состоянии
равновесия модуль сдвига равен модулю
вязкого трения ( ).
).

В наших условиях
 ,
,

 - уравнение
Гелемгольца-Смалуковского
- уравнение
Гелемгольца-Смалуковского
 - линейная скорость
смещении я фаз.
- линейная скорость
смещении я фаз.
E – напряженность электрического поля.
 - разность потенциалов
между обкладками
- разность потенциалов
между обкладками
 - электрофоретическая
подвижность [м 2 /(В*с)].
- электрофоретическая
подвижность [м 2 /(В*с)].
В модели Гелемгольца не учитывается тепловое движение молекул. Реально распределение ионов в двойном слое носит более сложный характер.
Гуи и Чапман выделили следующие причины возникновения ДЭС:
Переход иона из одной фазы в другую при установлении равновесия.
Ионизация вещества твердой фазы.
Достройка поверхности ионами, присутствующими в дисперсионной среде.
Поляризация от внешнего источника тока.
Двойной электрической слой имеет размытое или диффузное строение. Ионы стремятся равномерно распределиться во всем диффузном слое.
Диффузный слой состоит из противоинонв, протяженность слоя определяется их кинетической энергией. При температуре стремящейся к абсолютному нулю противоиноы максимально приближены к потенциалопределяющим ионам.
Даня теория базируется на двух уравнениях:
уравнение Больцмана

 - работа против
сил электростатического взаимодействия.
- работа против
сил электростатического взаимодействия.
 - объёмная плотность
заряда.
- объёмная плотность
заряда.

Уравнение Пуассона

Поскольку толщина
ДЭС много меньше размеров частицы и для
плоского ДЭС производная по координатам
 и
и упраздняется.
упраздняется.

Для е у при у<<1 функцию можно разложить в ряд Маклорена:

Ограничимся двумя членами ряда, тогда:


 -
толщина ДЭС – это расстояние, на котором
потенциал ДЭС уменьшается в e
раз.
-
толщина ДЭС – это расстояние, на котором
потенциал ДЭС уменьшается в e
раз.
Чем меньше
температура, тем меньше
 .
При Т→0 – плоский ДЭС. Чем больше
концентрация, тем большеI,
тем меньше
.
При Т→0 – плоский ДЭС. Чем больше
концентрация, тем большеI,
тем меньше
 .
.





 «–» означает, что
потенциал с расстоянием уменьшается.
=>
«–» означает, что
потенциал с расстоянием уменьшается.
=>
=>

 ,
,
 - потенциал экспоненциально уменьшается.
- потенциал экспоненциально уменьшается.
Потенциал для
поверхностной плотности заряда:

Поверхностный заряд – объемный заряд с противоположным знаком, проинтегрированный по расстоянию.




=>


Там, где потенциал
уменьшается в 2,7 раза -

Ёмкость двойного
слоя

Недостаток теории
– не учитывается наличие слоя Гелемгольца,
т.е. не учитывает
 ,
отсюда ошибки в определении основных
параметров. Также не объясняет влияние
ионов различной природы на толщину
двойного электрического слоя.
,
отсюда ошибки в определении основных
параметров. Также не объясняет влияние
ионов различной природы на толщину
двойного электрического слоя.
Теория Штерна. Строение коллоидной мицеллы.
Двойной электрический
слой состоит из двух частей: плотной и
диффузной. Плотный слой образуется в
результате взаимодействия потенциалобразующих
ионов со специфически адсорбирующимися.
Эти ионы, как правило, частично или
полностью дегидратированы и могут иметь
как одинаковый, так и противоположный
к потенциалопределяющим ионам заряд.
Это зависит от соотношения энергии
электростатического взаимодействия
 и потенциала специфической адсорбции
и потенциала специфической адсорбции .
Ионы плотного слоя закреплены. Другая
часть ионов расположена в диффузном
слое, эти ионы свободны и могут перемещаться
вглубь раствора, т.е. из области большей
концентрации в область меньшей. Общая
плотность заряда складывается из двух
частей.
.
Ионы плотного слоя закреплены. Другая
часть ионов расположена в диффузном
слое, эти ионы свободны и могут перемещаться
вглубь раствора, т.е. из области большей
концентрации в область меньшей. Общая
плотность заряда складывается из двух
частей.

 -заряд слоя
Гельмгольца
-заряд слоя
Гельмгольца
 -Заряд диффузного
слоя
-Заряд диффузного
слоя
Поверхность имеет определенное число адсорбционных центров, каждый из которых взаимодействует с одним противоионом. Константа такой квазихимической реакции равна:

 ,
где
,
где
 - мольная доля противоионов в растворе
- мольная доля противоионов в растворе
Распределение Гельмгольца
Потенциал убывает линейно
Распределение
потенциала по Гуи
.
Плотного слоя нет, потенциал убывает
экспоненциально со значения

Распределение по Штерну .
Вначале снижение потенциала линейно, а затем экспоненциально.
При наложении электрического поля в случае электрофореза движется не непосредственно частица твердой фазы, а частица твердой фазы со слоем окружающих её ионов. ДЭС повторяет форму частицы дисперсной фазы. При наложении потенциала отрывается часть диффузного слоя. Линия разрыва называется границей скольжения .
Потенциал,
возникающий на границе скольжения в
результате отрыва части диффузного
слоя, называется электрокинетическим
потенциалом
(Дзэта потенциал
 ).
).
Частица дисперсной фазы, с окружающим её слоем противоионов и двойным электрическим слоем называется мицеллой .
Правила написания коллоидных мицелл:


1-1 зарядный электролит
T – частица дисперсной фазы.
AA – граница плотной и диффузной части.
BB – граница скольжения.
Граница скольжения может совпадать с линией AA, а может и не совпадать.
Значения pH, при котором дзэта-потенциал равен нулю, называется изоэлектрической точкой .
CaCl 2 + Na 2 SO 4 → CaSO 4 ↓ + 2NaCl
1. В избытке CaCl 2
CaCl 2 ↔ Ca 2+ + 2Cl -
{CaSO 4 m∙nCa 2+ 2(n - x )Cl - } 2 x + x Cl - - запись мицеллы.
CaSO 4 m – агрегат.
CaSO 4 m∙nCa 2+ – ядро.
CaSO 4 m∙nCa 2+ 2(n - x )Cl - - частица.
2. В избытке Na 2 SO 4
Na 2 SO 4 ↔2Na + + SO 4 2-
{CaSO 4 m∙nSO 4 2- 2(n-x)Na + } 2x- 2xNa + - мицелла
CaSO 4 m – агрегат.
CaSO 4 m∙nSO 4 2 + – ядро.
CaSO 4 m∙nSO 4 2- 2(n-x)Na + - частица
Уравнение Гелемгольца-Смолуховского

 - линейная скорость
смещения границ (в электроосмосе).
- линейная скорость
смещения границ (в электроосмосе).
 - разность потенциалов
на обкладках конденсатора (в электроосмосе).
- разность потенциалов
на обкладках конденсатора (в электроосмосе).


 - объемная скорость
течения раствора, S
– площадь поперечного сечения ячейки.
- объемная скорость
течения раствора, S
– площадь поперечного сечения ячейки.

E – напряженность электрического поля.


 (для электроосмоса).
(для электроосмоса).
Для потенциала течения:

 - потенциал
- потенциал
 - давление на
мембрану
- давление на
мембрану
Как правило, значение электрофоретических подвижностей и электроосмотических подвижностей меньше расчетных. Это происходит вследствие:
Релаксационного эффекта (при движении частицы дисперсной фазы нарушается симметрия ионной атмосферы).
Электрофоретического торможения (возникновение дополнительного трения в результате движения противоионов).
Искажение линий тока в случае электропроводных частиц.
Связь поверхностного натяжения с потенциалом. Уравнение Липпмана.
Образование ДЭС происходит самопроизвольно вследствие стремления системы снизить свою поверхностную энергию. В условиях постоянства T и p обобщенное уравнение первого и второго законов термодинамики выглядит:
 (2)
(2)
(3), (1)=(3) =>
=>

 - 1-е уравнение
Липпмана.
- 1-е уравнение
Липпмана.
 - поверхностная
плотность заряда.
- поверхностная
плотность заряда.
 - дифференциальная
емкость.
- дифференциальная
емкость.
 - 2-е уравнение
Липпмана.
- 2-е уравнение
Липпмана.
С – емкость.
Решим 1-е уравнение Липпмана и фундаментальное уравнение адсорбции:
 ,
,


 ,
тогда
,
тогда
 - уравнение
Нернста
- уравнение
Нернста
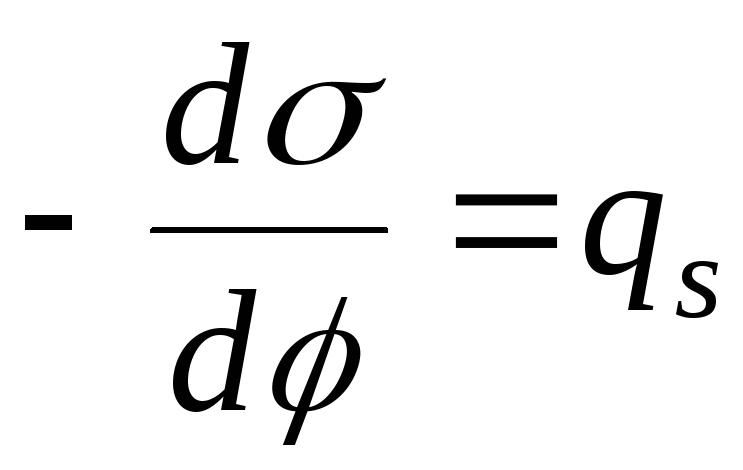 ,
,
 ,
,




 - уравнение
электрокапиллярной кривой (ЭКК).
- уравнение
электрокапиллярной кривой (ЭКК).
В
 :
: ,
но
,
но
Катионные ПАВы (КПАВ) снижают катодную ветвь ЭКК.
Анионные ПАВы (АПАВ) снижают анодную ветвь ЭКК.
Неионогенные ПАВы (НПАВ) снижают среднюю часть ЭКК.
Устойчивость дисперсных систем. Расклинивающее давление.
Дисперсные системы можно разделить:
Системы неустойчивые термодинамически могут быть устойчивы кинетически за счет перехода в метастабильное состояние.
Различают два вида устойчивости:
Седиментационная устойчивость (по отношению к силе тяжести).
Агрегативная устойчивость. (по отношению к слипанию)
Коагуляция – это процесс слипания частиц, приводящий к потере агрегативной устойчивости. Коагуляцию может вызвать изменение температуры, pH, перемешивание, ультразвук.
Различают коагуляцию:
Обратимая.
Необратимая.
Коагуляция протекает при введении электролитов.
Правила коагуляции:

Плёнка – это часть системы, находящаяся между двумя межфазными поверхностями.
Расклинивающее давление возникает при резком уменьшении толщины плёнки в результате взаимодействия сближающихся поверхностных слоев.

«-» - при уменьшении толщины пленки расклинивающее давление возрастает.
P 0 – давление в объемной фазе, которая является продолжением прослойки.
P 1 – давление в пленке.
Теория устойчивости. ДЛФО (Дерягин, Ландау, Фервей, Овербек).
Согласно теории ДЛФО в расклинивающем давлении выделяют две составляющие:
Электростатическая П Э (положительная, она обусловлена силами электростатического отталкивания). Соответствует уменьшению энергии Гиббса при возрастании толщины пленки.
Молекулярная П М (отрицательная, обусловлена действием сил притяжения). Обусловлена сжатием пленки за счет химических поверхностных сил, радиус действия сил десятые доли нм с энергией порядка 400 кДж/моль.
Полная энергия
взаимодействия
:

 - система агрегативно
устойчивая
- система агрегативно
устойчивая
 - неустойчивая
система
- неустойчивая
система
П оложительная
составляющая.
оложительная
составляющая.
Увеличение обусловлено увеличением потенциальной энергии при сжатии тонких пленок. Для пленок большой толщины избыточная энергия ионов скомпенсирована и равна энергетическому взаимодействию в объеме дисперсионной среды.
Если
 (
( - толщина пленки,
- толщина пленки, -
радиус иона) утоньшение пленки приводит
к исчезновению и уменьшению в ней молекул
и ионов с минимальной поверхностной
энергией. Число соседних частиц
уменьшается, в результате чего
потенциальная энергия оставшихся в
пленке частиц возрастает.
-
радиус иона) утоньшение пленки приводит
к исчезновению и уменьшению в ней молекул
и ионов с минимальной поверхностной
энергией. Число соседних частиц
уменьшается, в результате чего
потенциальная энергия оставшихся в
пленке частиц возрастает.


Теория ДЛФО взаимодействие частиц рассматривает как взаимодействие пластин.

Частицы не взаимодействуют



 - уравнение Лапласа,
- уравнение Лапласа,
 ,
,


Для слабо заряженных поверхностей
Для сильно заряженных поверхностей:

Молекулярная составляющая – взаимодействие двух атомов:
 ~
~
Взаимодействие атома с поверхностью:

Возьмем две пластинки:
Д ля
получения молекулярной составляющей
необходимо провести суммирование всех
энергий взаимодействия атомов правой
и левой пластин.
ля
получения молекулярной составляющей
необходимо провести суммирование всех
энергий взаимодействия атомов правой
и левой пластин.
где
 - постоянная Гамакера (учитывает природу
взаимодействующих тел).
- постоянная Гамакера (учитывает природу
взаимодействующих тел).
Т.о. энергия взаимодействия частиц в системе может быть выражена с помощью потенциальных кривых.
I – первичный потенциальный минимум. Это зона необратимой коагуляции, силы притяжения преобладают.
II – зона агрегативной устойчивости, преобладают силы отталкивания.
III – вторичный потенциальный минимум (или зона флокуляции). Между частицами дисперсной фазы существует прослойка электролита, и частицы могут быть разделены и переведены в зону агрегативной устойчивости.

Кривая 1 – система агрегативно устойчива.
Кривая 2 – в зоне I устойчива, в зоне II не устойчива.
Кривая 3 – в системе произошла коагуляция.
Кривая 4 – в точке
4 суммарная энергия взаимодействия U=0,
 ,
эта точка экстремума соответствует
началу быстрой коагуляции.
,
эта точка экстремума соответствует
началу быстрой коагуляции.
Существует два случая:
1. Поверхности слабозаряженные:
U = U Э + U M = 0
 (1)
(1)
2)

 (2)
(2)



 - это толщина
прослойки соответствующая началу
процессу коагуляции.
- это толщина
прослойки соответствующая началу
процессу коагуляции.






 - для слабозаряженных
поверхностей
- для слабозаряженных
поверхностей
 тогда
тогда

2. Для сильнозаряженных поверхностей:
 (1)
(1)
2)

 (2)
(2)


 (3)
(3)
 ,
,
Возведем (3) в квадрат

Коагуляция:
При специфической адсорбции ионы могут адсорбироваться в сверхэквивалентном количестве таким образом, что поверхность может изменить свой заряд. Происходит перезарядка поверхности.
В случае специфической адсорбции могут адсорбироваться ионы не только противоположных знаков, но и одного.
Если адсорбируются ионы того же знака, что и поверхность, то в поверхностном слое будет происходить не падение потенциала, а его рост.

Нейтрализационная коагуляция (протекает с участием слабозаряженных частиц и зависит не только от заряда электролита-коагулятора, но и от потенциала на границе плотного и диффузного слоя).
Теория быстрой коагуляции Смолуховского.

Зависимость скорости коагуляции от концентрации электролита.
I – скорость коагуляции мала,
II – скорость коагуляции практически пропорциональна концентрации электролита.
III – область быстрой коагуляции, скорость практически не зависит от концентрации.
Основные положения :
Исходный золь монодисперсный, сходные частицы имеют сферическую форму.
Все столкновения частиц результативны.
При столкновении двух первичных частиц образуется вторичная. Вторичная + первичная = третичная. Первичное, вторичное, третичное – кратность.
В терминах химической кинетики процесс коагуляции может быть описан уравнением:

Решением будет
уравнение:

 - время половинной
коагуляции. Это время, в течение которого
число частиц золя уменьшается в 2 раза.
- время половинной
коагуляции. Это время, в течение которого
число частиц золя уменьшается в 2 раза.

 ,
,
 ,
,
 ,
,


По мере увеличения
кратности максимум кривых коагуляции
сдвигается в сторону больших значений
 .
.
Недостатки:
Предположение о монодисперсности.
Предположение о результативности всех столкновений.
Адсорбция как самопроизвольное концентрирование молекул на поверхности сопровождается понижением энтропии системы. Так как критерием самопроизвольности процесса является
∆Н - T· ∆S = ∆G< 0,
то адсорбция возможна только при ∆Н < 0 (экзотермический процесс). Равновесие определяется условием ∆Н = T· ∆S. При повышении температуры равновесие смещается в сторону эндотермического процесса, т. е. десорбции.
Адсорбция на поверхности твердого тела
1. Мономолекулярная адсорбция.
По теории Ленгмюра молекулы адсорбтива взаимодействуют с поверхностью адсорбента, образуя в итоге мономолекулярный слой. B этом случае степень заполнения () поверхности адсорбируемым веществом при адсорбции из газовой фазы
из жидкости
где К - константа равновесия (константа адсорбции);
р - парциальное давление адсорбируемого газа;
с - концентрация адсорбируемого вещества.
Зависимость β от р (или с) представлена графиком (изотерма адсорбции, Т = const) на рис. 1.3.

Рис. 1.3. Степень заполнения поверхности адсорбируемым веществом
При малых концентрациях и парциальных давлениях адсорбция пропорциональна концентрации или парциальному давлению:
р<< 1, β ≈ К· р илис<< 1, β ≈ К· с, т.е. начальный участок изотермы приблизительно линеен, причем tg α = К(tg α определяют по наклону кривой при р (или с) → 0: или ).
Если - количество молей адсорбированного вещества на 1 г адсорбента; - максимально возможное количество молей адсорбированного вещества на 1 г адсорбента ("емкость монослоя"), то
Подставляя β в уравнение (1.3) (для случая адсорбции из газовой фазы концентрацию с в уравнениях следует заменить на давление р ), получаем:
 (1.6)
(1.6)
Так как и К в данной паре адсорбент-адсорбтив являются константами (при T =const), то по зависимости можно найти и К (рис. 1.4).

Рис. 1.4. Графическое решение уравнения адсорбции
получают путем экстраполяции экспериментальной линейной зависимости к () = 0; и, так как , то , .
Величину можно использовать для определения удельной поверхности адсорбента УД (в м 2 на 1 г адсорбента), если известна площадь ω, занимаемая на поверхности одной молекулой адсорбтива (определяется из размеров молекулы):
УД = · ω · Nа, (1.7)
где Nа - число Авогадро (Nа = 6,02 · 10 23).
В свою очередь, известную величину УД можно использовать для расчета или ωлюбого вещества по его адсорбции на данном адсорбенте.
2. Полимолекулярная адсорбция.
Уравнение (1.5) описывает кривую с насыщением, т.е. при
р (или с) → ∞ стремится к предельному значению, равному (рис. 1.5,а).

Рис.1.5. Изотермы адсорбции:
а – адсорбция с насыщением; б – полимолекулярная адсорбция
Однако в некоторых случаях изотермы адсорбции выглядят как показано на рис. 1.5,б, т.е. не достигает предела даже при высоких р (или с).
Зависимости типа показанной на рис. 1.5,б соответствуют полимолекулярной адсорбции. Как правило, такие изотермы характерны для веществ с сильными межмолекулярными взаимодействиями (например, для воды). Когда центры адсорбции на поверхности адсорбента заняты (мономолекулярный слой насыщен), "посадка" следующих молекул адсорбата происходит за счет межмолекулярных взаимодействий с уже адсорбированными молекулами (рис.1.6). Теплота такой адсорбции близка по абсолютной величине, но противоположна по знаку теплоте испарения соответствующей жидкости (подумайте, почему).

Рис.1.6. Схема адсорбции:
а - мономолекулярная адсорбция; б - полимолекулярная адсорбция
По мере приближения р к давлению насыщенного пара адсорбируемого вещества оно начинает конденсироваться на поверхности адсорбента, в результате быстро растет с ростом р .
Основные определения и способы классификации адсорбционных процессов.
Адсорбция относится к явлениям, происходящим вследствие самопроизвольного уменьшения поверхностной энергии.
Адсорбция – процесс самопроизвольного обратимого или необратимого перераспределения компонентов гетерогенной системы между поверхностным слоем и объемом гомогенной фазы.
В многокомпонентных системах в поверхностный слой предпочтительнее переходит компонент, который сильнее снижает межфазное натяжение. В однокомпонентных системах при формировании поверхностного слоя происходит изменение его структуры (определенная ориентация атомов и молекул, поляризация), называемое автоадсорбцией .
Более плотную фазу, на которой локализованы адсорбционные взаимодействия называют адсорбентом . Вещество, перераспределяемое между объемом гомогенной фазы и поверхностным слоем, обозначают термином «адсорбат ».
В ряде случаев процесс адсорбции является обратимым. В этом случае при определенных условиях часть адсорбированных молекул в результате молекулярно-кинетических явлений может перейти из поверхностного слоя в объем фазы. Процесс, обратный адсорбции, называют десорбцией .
Способы классификации адсорбционных процессов.
Классификация адсорбционных процессов по агрегатному состоянию взаимодействующих фаз. В зависимости от агрегатного состояния смежных фаз различают следующие типы адсорбционных процессов:
Адсорбция газов на твердых адсорбентах;
Адсорбция растворенных веществ на границах раздела «твердое тело – жидкость» и «жидкость – жидкость»;
Адсорбция поверхностно-активных веществ на границе раздела «жидкость – газ».
Классификация адсорбционных процессов по механизму взаимодействия адсорбента и адсорбата. Адсорбцию можно рассматривать как взаимодействие молекул адсорбата с активными центрами адсорбента. По механизму их взаимодействия подразделяют следующие виды адсорбции:
1) физическая (молекулярная) адсорбция – взаимодействие между молекулами адсорбата и адсорбента осуществляется за счет сил Ван-дер-Ваальса, водородных связей (без протекания химических реакций);
2) химическая адсорбция (хемосорбция) – присоединение молекул адсорбата к активным центрам адсорбента происходит в результате протекания химических реакций различных типов (за исключением реакций ионного обмена);
3) ионообменная адсорбция (ионный обмен) – перераспределение вещества адсорбата между раствором и твердой фазой (ионитом) по механизму реакций ионного обмена.
Для количественного описания адсорбционных процессов применяют две величины.
1) Абсолютная адсорбция – количество (моль) или масса (кг) адсорбата на единицу площади поверхности или массы адсорбента. Обозначение – А; размерность: моль/м 2 , моль/кг, кг/ м 2 , кг/кг.
2) Гиббсовская (избыточная) адсорбция – избыток вещества адсорбата в поверхностном слое определенной толщины по сравнению с его количеством в объеме гомогенной фазы, отнесенный к единице площади поверхности или массы адсорбента. Обозначение – Г; размерность: моль/м 2 , моль/кг.
Связь между абсолютной и избыточной адсорбции можно проиллюстрировать с помощью уравнения:
Г = А – с * h (3.1)
где с – равновесная концентрация вещества в объеме фазы, моль/м 3 ;
h - толщина поверхностного слоя, условно принимаемая равной 10 -9 м.
В многокомпонентных гетерогенных системах при перераспределении того или иного компонента между объемом гомогенной фазы и поверхностным слоем справедливо уравнение для избыточной внутренней энергии поверхности:
U = T * S + s * s + Sm i * n i (3.2)
Приведя все члены уравнения к единице площади межфазной поверхности, получим:
U s = T * S s + s + Sm i * Г i (3.3)
где Г i = n i / s – избыток i -го компонента в поверхностном слое, то есть гиббсовская адсорбция.
Для однокомпонентной системы уравнение (3.3) примет вид:
G s = s + m * Г (3.4)
где G s = U s - T * S s – энергия Гиббса поверхности или работа создания единицы площади поверхности;
m * Г – уплотнение вещества адсорбируемого вещества в поверхностном слое.
Исходя из уравнения (3.4) можно сделать вывод о том, что при адсорбции работа по созданию межфазной поверхности складывается из работы образования поверхности (разрыва когезионных связей в объеме фазы адсорбата) и уплотнения вещества в поверхностном слое.
В состоянии динамического равновесия между адсорбентом и адсорбатом изменение энергии Гиббса гетерогенной системы ΔG = 0, термодинамика процесса адсорбции описывается уравнением, получившим название фундаментальное адсорбционное уравнение Гиббса :
Ds = SГ i * dm i (3.5)
Данное уравнение является универсальным, так как справедливо для всех типов адсорбционных процессов
Частные случаи адсорбционного уравнения Гиббса.
1) Адсорбция из растворов.
Для химического потенциала i -го компонента системы при протекании адсорбции на границах раздела «жидкость – твердый адсорбент» и «жидкость – газ» справедливы уравнения:
m i = m i 0 + R*T*ln a i (3.6)
dm i = R*T* d ln a i (3.7)
где m i 0 - химический потенциал i -го компонента системы при стандартных условиях;
a i – активность i -го компонента системы при стандартных условиях.
Исходя из этого, адсорбционное уравнение Гиббса примет вид:
Г i = - a i / R*T * (ds / da i) (3.8)
Для растворов неэлектролитов принимаем a i = с i , тогда:
Г i = - с / R*T * (ds / dс) (3.9)
Для растворов электролитов:
Г i = - с ± n / R*T * (ds / dс ± n) (3.10)
где с ± - средняя ионная концентрация раствора;
n - стехиометрический коэффициент.
2) Адсорбция веществ из газовой фазы.
В соответствии с уравнением Менделеева-Клайперона:
Р = с * R*T (3.11)
В связи с этим, уравнение Гиббса для адсорбции газов на твердых адсорбентах записывают в следующей форме:
Г i = - Р / R*T * (ds / dР) (3.12)
На практике адсорбционное уравнение Гиббса позволяет по данным измерения поверхностного натяжения при различных значениях концентрации жидкости или равновесного давления газа рассчитать величину адсорбции веществ в межфазном слое, для которого определено поверхностное натяжение.
Адсорбция
(от лат. ad - на, при и sorbeo - поглощаю), изменение (обычно - повышение) концентрации вещества вблизи поверхности раздела фаз ("поглощение на поверхности"). Причина адсорбции
- ненасыщенностью межмолекулярных связей вблизи поверхности, т.е. существованием адсорбционного силового поля. Тело, создающее такое поле, называют адсорбентом, вещество, молекулы которого могут адсорбироваться, - адсорбтивом, уже адсорбированное вещество - адсорбатом. Процесс, обратный адсорбции
, называется десорбцией.
Природа адсорбционного поля различна. В случае, если адсорбция связна с ван-дер-ваальсовыми связями, то адсорбцию
называют физической. Если это валентные связи, т.е. адсорбция
проходит с образованием поверхностных химических соединений, то адсорбцию
называют химической, или хемосорбцией
. Важными чертами хемосорбции
являет: необратимость, высокие тепловые эффекты (сотни кДж/моль), активированный характер. Существует множество промежуточных видов адсобрции
между физической и химической адсорбцией
. Например, адсорбция
, вызванная образованием водородных связей. Так же возможны разные виды физической адсорбции
. Наиболее часто встречается возникновение дисперсионных межмолекулярных сил притяжения, в связи с тем, что они приблизительно постоянны для адсорбентов с поверхностью любой химической природы (неспецифическая адсорбция
). Физическая адсорбция
может быть вызвана электростатическими силами (взаимодействием между ионами, диполями или квадруполями); при этом адсорбция
определяется химической природой молекул адсорбтива (так называемая специфическая адсорбция
). Важную роль играет также геометрия поверхности раздела. если поверхность является плоской, то это адсорбция
открытой поверхности, в случае слабо или сильно искривленной поверхности - об адсорбции
в порах адсорбента.
В теории адсорбции
различают статику (система адсорбент-адсорбат находится в термодинамическом равновесии) и кинетику (равновесия нет).
Статика адсорбции
Термодинамика адсорбции
.Основы термодинамики адсорбции
были созданы Дж.Гиббсом в 70-е гг. XIX в. По Гиббсу, в равновесной двухфазной системе вблизи поверхности раздела фаз происходит некоторое изменение локальных значений всех экстенсивных свойств (кроме объема). Однако фазы считаются однородными вплоть до некоторой геометрической поверхности, разделяющей их. Поэтому значение какого-либо экстенсивного свойства для системы в целом не равно сумме значений этого свойства в однородных фазах и . Разность приписывается двухмерной поверхностной фазе, связанной с разделяющей поверхностью. Т.к. поверхностная фаза не имеет толщины, то V 0
=+ и =-, где V-
объем.
Изложенные представления позволяют привести фундаментальное термодинамическое уравнение к виду:
где G-гиббсова свободная энергия, S-энтропия, - межфазное поверхностное натяжение, s-площадь поверхности раздела, и n i - соответствующий химический потенциал и число молей i -того компонента. Индекс указывает на значение соответствующего свойства в поверхностной фазе. Преобразование Лежандра позволяет видоизменить уравнение (1) для изотермических условий:
Величина называется гиббсовой адсорбцией и обозначается символом Г, (выражается в моль/см 2). Для двух-компонентной системы:
Положение разделяющей поверхности может быть выбрано произвольно. В частности, выбор этого положения может удовлетворять условию Г 1 =0. Такая поверхность называется эквимолекулярной. Для нее вводится обозначение Г 2 = Г 2 (1) . Отсюда следует основное адсорбционное уравнение Гиббса:
Если адсорбтив совершенно не растворим в одной из двух фаз, =const, и переход от уравнения (2) к уравнению (3) не требует условия Г 1 =0. Таким образом, гиббсова адсорбция - это избыток данного компонента в реальной двухфазной системе по сравнению с такой системой, в которой обе фазы были бы строго однородны вплоть до разделяющей поверхности. Кроме гиббсовых избыточных величин адсорбции , в ее теории большую роль играет адсорбция , понимаемая как полное содержание компонента i в пространстве W , в котором проявляются адсорбционные силы. Обозначая полное содержание через а и считая, что компонент i совершенно не растворим в одной из объемных фаз, имеем:
где c i -концентрация i -того компонента в объемной фазе. При малых с i :
Адсорбция
может происходить на любой поверхности раздела между двумя любыми фазами, в частности на поверхности раздела флюид-флюид (жидкость-газ, жидкость-жидкость) или твердое тело-флюид (твердое-газ, твердое-жидкость). В системах флюид-флюид можно измерить а как функцию и экспериментально определить Г 2 (1) по уравнению (3). Во втором случае дпя определения Г 2 (1) измеряют любым методом n i
0 , ,
и концентрации i-того компонента в этих объемах. Отсюда вычисляют Г i
(1) . Этот метод называется объемным (или волюмометрическим). При весовом (гравиметрическом) методе непосредственно определяют количество i
-того компонента на поверхности раздела.
Изотерма адсорбции
.В равновесной адсорбционной системе параметры, определяющие равновесие, - это a i парциальные давления р (или с i ) и температура Т . Они связаны так называемым термическим уравнением:
При адсорбции индивидуального адсорбтива (i =1) это уравнение принимает вид:
Три частных случая термического уравнения (когда Т, р или a - константы) играют особую роль в теории адсорбции :
а= - уравнение изотермы адсорбции ,
Т= - уравнение изобары адсорбции ,
Р-
- уравнение изостеры адсорбции
.
Конкретный вид функций и определяется особенностями рассматриваемой системы. Если одна из них, например, известна для любого значения Т=
const, то, очевидно, становятся известными и две другие. При этом не обязательно, чтобы был известен аналитический вид зависимостей. Они могут быть заданы эмпирически в виде набора значений а, р
и Т
.
В теории адсорбции
обычно решается вопрос о виде функции а
=(р) г, т.е. об уравнении изотермы адсорбции
. Эта проблема связана с тепловыми эффектами, сопровождающими адсорбцию
. При расчете изменения значений основных термодинамических функций в случае перехода dn
молей адсорбтива из объемной фазы в поверхностную в равновесной системе при р =
const возможны два случая: в первом учитывается только превращение адсорбтива в адсорбат, поскольку адсорбент при адсорбции
термодинамически неизменен и его роль - служить источником адсорбционного поля; во втором учитывается и изменение адсорбента.
Так как система равновесна, то химические потенциалы адсорбата и адсорбтива одинаковы; энтропия адсорбата вследствие уменьшения подвижности молекул при адсорбции
меньше энтропии адсорбтива. Поэтому при инертном адсорбенте энтальпия всегда отрицательна, т.е. адсорбция
экзотермична. Учет изменения энтропии адсорбента может изменить этот вывод. Например, при сорбции полимерами веществ, в которых полимер набухает, энтропия последнего (из-за увеличения подвижности макромолекул) может столь сильно возрасти, что адсорбция
становится эндотермической. В дальнейшем в статье рассматривается только экзотермическая адсорбция
.
Различают интегральную, дифференциальную, изостерическую и среднюю теплоты адсорбции
. Интегральная теплота Q
равна убыли энтальпии (при V=
const - постоянной внутренней энергии) при изменении адсорбции
от a 1
до а 2
(в частном случае может быть а 1 =0): Q= -(Н 2 - Н 1
). Эту величину относят обычно к массе адсорбента и выражают в Дж/кг.
Дифференциальная теплота q (Дж/моль) равна убыли энтальпии dH при изменении а на da . Ее выражают отношением q = - (dH/da) . Очевидно, что
Изостерическую теплоту q st принимают равной:
где - разность мольных объемов адсорбата и адсорбтива. Можно показать, что  для идеального газового адсоритива:
для идеального газового адсоритива:
Смысл введения q si в том, что для ее определения не требуется калориметрических данных (таких, как Q и q ) и она может быть вычислена по уравнению (9) по результатам измерения адсорбции . Вводят также среднюю теплоту Q (Дж/моль):
С ростом а
параметр Q
всегда возрастает, a q
может уменьшаться, увеличиваться или оставаться неизменной. С ростом а
при
неоднородной поверхности адсорбция
происходит на все менее активных участках, что приводит к уменьшению q
. Однако при этом уменьшаются средние расстояния между адсорбированными молекулами, вследствие чего увеличиваются силы притяжения между ними, и q
возрастает. Соотношение между двумя упомянутыми эффектами определяет ход зависимости q=f(a)
. При очень больших а
начинают преобладать силы отталкивания и в этой области q
всегда снижается с ростом a
.
При очень малых заполнениях поверхности уравнение изотермы адсорбция имеет вид уравнения Генри:
где К H - коэффициент Генри. Действительно, при очень малых а адсорбционный слой подобен двухмерному идеальному газу, поэтому его уравнение состояния имеет вид: =RT, где - двухмерное давление, - площадь, занимаемая одним молем вещества. Отсюда, учитывая, что =-, и используя уравнение (3), получаем уравнение (12). Уравнение Генри требует, чтобы q было постоянным. При больших заполнениях это уравнение перестает выполняться. Поэтому Г.Фрейндлих (1906) предложил описывать изотермы адсорбции следующим эмпирическим уравнением (уравнение Фрёйндлиха):
где k
и n
- константы. Этим уравнением часто пользуются как интерполяционной формулой, хотя оно при малых р
не переходит в уравнение (12), а при очень больших р
приводит к несогласующемуся с опытом неограниченному возрастанию а
.
Строгая теория изотермы адсорбция была создана И. Ленгмю-ром (1914-18). В основу теории положена след. модель: 1) поверхность адсорбента представляет собой набор энергетически одинаковых активных центров, на которых адсорбируются (локализуются) молекулы адсорбтива; 2) на одном центре адсорбируется только одна молекула, т.е. при адсорбция образуется только один адсорбц. слой (монослой); 3) адсорбция на данном центре не влияет на адсорбция на др. центрах, т. е. взаимод. адсорбированных молекул можно пренебречь.
Модель Ленгмюра наз. локализованной мономолекулярной адсорбция на однородной поверхности. уравнение изотермы адсорбция , соответствующее этой модели, м.б. получено при помощи разл. методов (молекулярно-кинетич., термодинамич., ста-тистико-термодинамич.). Так, адсорбц. равновесие можно выразить след. схемой:
Молекула Своб. Адсорбц. в газовой + адсорбц. комплекс фазе центр (занятый центр)
Концентрация молекул в газе пропорциональна р, концентрация своб. центров-величине (а т - а), где а т - полное число центров, а-число занятых центров, концентрация адсорбц. комплексов-величине адсорбция Следовательно, константа равновесия равна: К р = р(а т - а)/адсорбция Отсюда получаем уравнение Ленгмюра:
где b -т. наз. адсорбц. коэф., равный К р -1 . В области очень малых давлений bр " 1 и a = (a m b)p, что отвечает уравнению Генри, в котором К H = a m b. В области очень больших давлений bр 1 и аа т; при этом адсорбция перестает зависеть от давления. Константа равновесия b -1 связана со стандартным значением изобарного потенциала реакции:
Модель Ленгмюра требует, чтобы дифференц. теплота и энтропия адсорбция не зависели от степени заполнения поверхности.
уравнение (14)-строгое выражение, соответствующее модели Ленгмюра, однако оно редко оправдывается на опыте, поскольку сама модель идеализированадсорбция Учение об адсорбция с 20-х гг. 20 в. в значит. степени строилось на основе ослабления или исключения того или иного допущения Ленгмюрадсорбция
Уже Ленгмюр предложил способ описания адсорбция на неоднородной поверхности (т.е. при допущении, что не все центры оди наковы). Объединяя одинаковые центры в группы и полагая, что к каждой группе применимо уравнение (14), можно считать, что адсорбция на всей поверхности выражается суммой членов уравнения (14):
Полагая, что число адсорбц. центров м.б. описано непрерывной ф-цией распределения по значениям своб. энергии, Я.Б.Зельдович получил из ф-лы (16) для экспоненциальной ф-ции уравнение типа (13).
адсорбция на неоднородных поверхностях-большая глава теории адсорбция Ее осн. задача-решение интегрального уравнения:
где f(р )- т. наз. эмпирич. изотерма адсорбция , -та или иная ф-ция распределения числа центров по значениям своб. энергии,(b, р)- локальная изотерма адсорбция , в кач-ве которой обычно принимают изотерму Ленгмюрадсорбция
Много попыток сделано в направлении отказа от второго допущения Ленгмюрадсорбция На этом пути особое значение приобрела теория полимолекулярной адсорбция , предложенная С. Брунауэром, П. Эмметом и Э. Теллером (теория БЭТ). Теория постулирует, что при температуре ниже критической каждая молекула, адсорбированная в первом слое (теплота адсорбции q i ,), является центром для молекул, образующих второй слой, и т.д. При этом считается, что теплота адсорбция во всех слоях, кроме первого, равна теплоте конденсации Такая модель приводит к уравнению:
где с = ехр[(q 1 -)/RT]. уравнение (18) в координатах a, p/p s соответствует S-образной кривой. В координатах p/p s ,
изотерма адсорбция по уравнению (18) должна быть линейной. Наклон этой прямой (обычно в интервале 0,05 p/p s 0,30) и отрезок, отсекаемый ею на оси ординат, дают значения соотв. а т и с. Широкое распространение теории БЭТ связано с тем, что ее авторы, фактически считая адсорбция нелокализованной, отождествляют константу а т не с числом дискретных адсорбц. центров, а с числом молекул адсорба-та в первом слое при плотнейшей упаковке (при р = p s). Поэтому, вводя представление о площади занимаемой одной молекулой в этом слое, принимают:
где s- площадь поверхности адсорбатадсорбция Как правило, для этого измеряют изотерму адсорбция азота и принимают, что для его молекулы= 0,162нм 2 . Часто выполняемый аналогичный расчет s по модели Ленгмюра не корректен, т.к. этот метод, очевидно, применим только к нелокализованной адсорбция
В теорию полимолекулярной адсорбция большой вклад внес Я. де Бур, экспериментально показавший, что зависимость среднего числа слоев (свыше первого) на всех поверхностях, близких по хим. природе, от p/p s выражается универсальной кривой (т. наз. t-кривой). Это также позволяет оценивать площади поверхности адсорбтивов.
Предпринимались попытки учесть в модели Ленгмюра также взаимод. между адсорбиров. молекулами. Так, Т. Хилл и Я. де Бур, считая, что уравнение состояния адсорбц. слоя есть двухмерный аналог уравнения Ван-дер-Ваальса, получили след. уравнение изотермы адсорбция :
где= а/а т, а и b-константы уравнения Ван-дер-Ваальсадсорбция Р. Фаулер и Э. Гуггенгейм, учтя взаимод. адсорбиров. молекул, вывели уравнение:
где-константа, связанная с парным взаимодействием молекул.
Существует еще один механизм, приводящий к дополнит. адсорбция адсорбтивов ниже их критич. температуры на пористых адсорбентах при сравнительно высоких значениях p/p s . Это - капиллярная конденсация. Если в поре образовался вогнутый мениск адсорбата, то в ней начинается конденсация при p/p s Согласно уравнению Кельвина:
где-поверхностное натяжение адсорбата, V-его мольный объем, r-радиус кривизны менискадсорбция Капиллярная конденсация приводит к резкому подъему изотермы адсорбция При этом часто (но не всегда) наблюдается т. наз. адсорбц. гистерезис, т.е. несовпадение адсорбц. и десорбц. ветвей изотермы. Как правило, это связано с тем, что формы менисков при адсорбция и десорбции не совпадают.
Капиллярную конденсацию используют для определения размеров пор адсорбентадсорбция По уравнению (22) для каждого значения p/p s вычисляют радиус кривизны менискадсорбция Из него, учитывая толщину адсорбц. слоя (напр., по t-кривой), форму переходной области от слоя к мениску и зависимость от кривизны при очень малых r, находят линейный размер (эффективный радиус r ef) пор, заполняемых при данном p/p s . Объем таких пор определяют по приросту адсорбция в этой точке изотермы. Используя полученные данные, строят кривую распределения объема пор по их радиусам. Метод применим при r ef 1,5 нм. Обычно расчет ведут по десорбц. ветви изотермы, но более строгая совр. теория требует для построения кривой учета обеих ветвей.
Потенциальная теория адсорбции и теория объемного заполнения микропор. Модель адсорбция , принципиально отличную от ленгмюровской, предложил в 1914 М. Поляки. Согласно этой модели, вблизи поверхности адсорбента существует потенциальное адсорбц. силовое поле, убывающее с расстоянием от поверхности. Вследствие этого давление адсорбтива, равное вдали от поверхности р, вблизи нее возрастает и на некотором расстоянии достигает значения p s , при котором адсорбтив конденсируется. Объем слоя между поверхностыо раздела и геом. местом точек, где р = p s , заполнен жидкостью, которой приписываются нормальные значения физ. свойств объемной жидкости. Обратимая изотермич. работа е адсорбц. сил, определяемая по уравнению= RTlnp/p s , наз. адсорбц. потенциалом, а вся концепция-потенциальной теорией адсорбция При заданной величине объема V адсорбц. слоя потенциалне зависит от температуры (вследствие независимости дисперсионных сил от температуры). Такая температурная инвариантность дает возможность пересчитывать адсорбция с одной т-ры на другую, хотя уравнения изотермы адсорбция на основе излагаемой теории вывести не удавалось. Модель Поляни широко и успешно применялась мн. авторами, однако она содержала два очень уязвимых положения: 1) допущение о том, что тончайшая адсорбц. пленка имеет нормальные значения физ. свойств объемной жидкости (это допущение не подтверждалось опытами); 2) температурная инвариантность ф-ции=f(V), лежащая в основе теории, приближенно подтверждалась опытом только для очень тонкопористых адсорбентов.
Используя потенциальную теорию, М.М. Дубинин предложил и разработал теорию объемного заполнения микро-пор (ТОЗМ). Было постулировано, что эта теория применима только к микропористым адсорбентам. Особенность таких адсорбентов, в которых линейные размеры пор r1 нм, состоит в том, что весь объем их пор "заполнен" адсорбц. полем. Поэтому при адсорбция они заполняются не послойно, а объемно. Величина в рассматриваемом случае - это не адсорбц. потенциал, а с точностью до знака хим. потенциал адсорбата, отсчитываемый от уровня хим. потенциала нормальной жидкости при той же температуре. Вся совокупность пор адсорбентов разделяется на три класса: микропоры (r 0,6 нм), мезопоры (0,6 нмr20 нм) и макропоры (r 20 нм). адсорбция в микропорах происходит по схеме ТОЗМ, т.е. объемно, в мезопорах-по механизму послойного заполнения, завершаемого капиллярной конденсацией. Макропоры при адсорбц. равновесии никакой роли не играют.
Введя представление о ф-ции распределения объемов пор по значениям хим. потенциала адсорбата в них, М.М. Дубинин и Л. В. Радушкевич получили уравнение изотермы адсорбции ТОЗМ, которое обычно записывают в след. форме:

где п, Е и а 0 -параметры (а 0 = а при р = p s). Температурная зависимость a 0:
где= -(da 0 /dT); a 0 0 = a 0 при Т= Т 0 . Параметры п и Е практически не зависят от температуры. В большинстве случаев п = 2. Лишь для случаев, когда начальные теплоты адсорбция очень велики, п > 2. Для пересчета изотерм адсорбция с одного адсорбтива на другой приближенно допускают, что E 1 /E 2 P 1 /P= и что a 01 /a 02 V 1 /V 2 ,где P i -парахор, V i - мольный объем адсорбтивадсорбция
Каждый микропористый адсорбент характеризуется по ТОЗМ двумя параметрами: W- объемом микропор (W 0 = = a 0 V 0 )и E 0 -характеристич. энергией; W 0 и E 0 относят к стандартному адсорбтиву, обычно к бензолу.
Пользуясь представлением, что в реальном адсорбенте имеются поры разных размеров, и вводя распределение значений Е с дисперсией, равной Ф. Стекли предложил обобщение уравнения (23), названное уравнением Дубинина-Стёкли:

где B 0 -
константа, связанная с E
в уравнении
(23), а у=![]() .
T.к. в адсорбц. технике наиб. распространение получили именно микропористые
адсорбенты (активные угли, цеолиты, тонкопористые ксерогели), ТОЗМ применяется
не только в физ.-хим. исследованиях, но и в инженерных расчетах.
.
T.к. в адсорбц. технике наиб. распространение получили именно микропористые
адсорбенты (активные угли, цеолиты, тонкопористые ксерогели), ТОЗМ применяется
не только в физ.-хим. исследованиях, но и в инженерных расчетах.
Адсорбция газовых и жидких смесей. Па практике всегда имеют дело не с индивидуальным адсорбтивом, а со смесью газов или с жидкими растворами. Поэтому требуется обобщение теории адсорбция на случай многокомпонентного адсорбтивадсорбция В принципе можно исходить из любой модели адсорбция и распространить ее на этот случай. При адсорбция газовой смеси это достигается не только большим усложнением уравнений, но и введением в них дополнит. эмпирич. параметров, связанных или с взаимод. разнородных молекул или, в более общем виде, с влиянием одних в-в на коэф. активности других. Только модель Ленгмюра позволяет получить уравнение изотермы адсорбция смеси без параметров, не входящих в уравнения для адсорбция индивидуальных в-в. Для этого достаточно учесть, что при адсорбции k-того компонента из смеси i компонентов часть адсорбц. центров м.б. занята др. молекулами. Поэтому:
В случае адсорбция жидких растворов независимо от их концентрации вся поверхность адсорбента заполненадсорбция Вследствие этого адсорбция молекулы k-того компонента сопровождается вытеснением не-которого числа молекул остальных компонентов, т. е. адсорбция носит конкурентный характер.
Различают молекулярную и ионную адсорбция растворов. Первая происходит при адсорбция растворов неэлектролитов, вторая-р-ров электролитов. Молекулярная адсорбция , как правило, выражается избыточными величинами. Конкурентный характер адсорбция обусловливает то, что величина а м.б. как положительной, так и отрицательной. Выражая адсорбция i -того компонента как ф-цию его мольной доли в растворе х i -, имеем, что Г i = О при х i = 0 и х i = 1 (возможным изменением объема вещества в адсорбц. слое пренебрегают). Поэтому изотерма адсорбция имеет один или неск. экстремумов.
уравнение изотермы адсорбция бинарных растворов неэлектролитов, надежно обоснованное термодинамически, имеет вид:

где индекс s указывает на адсорбц. фазу, - (dn s 2 /dn s 1 )показывает, сколько молей второго компонента вытесняется одним молем первого,-разность слагаемых (стандартных частей) хим. потенциала, зависящих только от температуры.
Осн. проблема использования этого и ряда др. уравнений изотермы адсорбция -выяснение зависимости коэф. активности компонентов в адсорбц. слое от его составадсорбция Важнейший вопрос при применении адсорбция для разделения или очистки веществ-подбор селективного адсорбента по отношению к данному компоненту растворадсорбция
Ионная адсорбция , как правило, не носит эквивалентного характерадсорбция На поверхности из раствора электролита адсорбируются преим. катионы или анионы. Благодаря электрич. (кулонов-ским) силам на поверхности образуется двойной электрический слой.
Если в состав адсорбента входят ионы или поверхностные функц. группы, способные в данном растворителе к ионизации, то между адсорбентом и раствором электролита происходит ионный обмен. Адсорбент в этом случае наз. ионитом.
Кинетика адсорбции
адсорбция , как и любой реальный процесс, происходит во времени. Поэтому полная теория адсорбция должна содержать раздел о кинетике адсорбция Элементарный акт адсорбция осуществляется практически мгновенно (исключение-хемосорбция). Поэтому временные зависимости адсорбция определяются в осн. механизмом диффузии, т. е. подвода адсорбтива к месту адсорбция Если адсорбция на открытой поверхности не мгновенна, такой процесс происходит во внешнедиффузионной области; при этом законы диффузии не специфичны для адсорбция В случае же пористых адсорбентов, кроме внеш. диффузии, важную роль начинает играть внутр. диффузия, т.е. перенос адсорбтива в порах адсорбента при наличии в них градиента концентрации. Механизм такого переноса может зависеть от концентрации адсорбтива и размеров пор.
Различают молекулярную, кнудсеновскую и поверхностную (фольмеровскую) диффузию. Молекулярная диффузия осуществляется, если длина своб. пробега молекул в порах меньше размера пор, кнудсеновская-если эта длина превышает размер пор. При поверхностной диффузии молекулы перемещаются по поверхности адсорбента без перехода в объемную фазу. Однако значения коэф. диффузии не одинаковы для разных механизмов диффузии. Во мн. случаях экспериментально не удается установить, как именно происходит диффузия, и поэтому вводят т. наз. эффективный коэф. диффузии, описывающий процесс в целом.
Осн. эксперим. материалом о кинетике адсорбция служит т. наз. кинетич. кривая, т.е. ф-ция= а/а равн =f(t ) где-относительная адсорбция , равная отношению текущего значения адсорбции а к a равн - её значению при времени t. Для истолкования кинетич. кривой в простейшем случае предполагают, что зерно адсорбента имеет совершенно однородную по объему пористую структуру (эту модель наз. квазигомогенной). значит. усовершенствование квазигомогенной модели-представление о том, что каждое зерно содержит области с более крупными и более тонкими порами. Диффузия в таком зерне описывается двумя разл. коэффициентами.
В случае открытой поверхности, принимая модель Ленгмюра, легко получить кинетич. уравнение адсорбция Скорость приближения к равновесию представляет собой разность скоростей адсорбция и десорбции. Считая, как обычно в кинетике, что скорости процессов пропорциональны концентрациям реагирующих в-в, имеем:
где k адс и k дес - константы скорости соотв. адсорбция и десорбции. Давление в газовой фазе считается постоянным. При интегрировании этого уравнения от t = 0 до любого значения t получим:
Отсюда при f имеем:= равн. Поэтому окончательно имеем:
где k = k адс + k дес.
Влияние температуры на скорость адсорбция выражается уравнением, аналогичным уравнению Аррениусадсорбция С увеличением температуры k адс экспоненциально возрастает. Т.к. диффузия в порах адсорбента связана с преодолением активац. барьеров, температурные зависимости k адс и k дес не одинаковы.
Знание скоростей диффузии важно не только для теории адсорбция , но и для расчета пром. адсорбц. процессов. При этом обычно имеют дело не с отдельными зернами адсорбента, а с их слоями. Кинетика процесса в слое выражается очень сложными зависимостями. В каждой точке слоя в данный момент времени величина адсорбция определяется не только видом уравнения изотермы адсорбция и закономерностями кинетики процесса, но также аэро- или гидродинамич. условиями обтекания зерен газовым или жидкостным потоком. Кинетика процесса в слое адсорбента в отличие от кинетики в отдельном зерне наз. динамикой адсорбция , общая схема решения задач которой такова: составляется система дифференц. уравнений в частных производных, учитывающая характеристики слоя, изотерму адсорбция , диффузионные характеристики (коэф. диффузии, виды переноса массы по слою и внутри зерен), аэро- и гидродинамич. особенности потокадсорбция Задаются начальные и краевые условия. Решение этой системы уравнений в принципе приводит к значениям величин адсорбция в данный момент времени в данной точке слоя. Как правило, аналитич. решение удается получить только для простейших случаев, поэтому такая задача решается численно с помощью ЭВМ.
При опытном изучении динамики адсорбция через слой адсорбента пропускают газовый или жидкостный поток с заданными характеристиками и исследуют состав выходящего потока как ф-цию времени. Появление поглощаемого вещества за слоем наз. проскоком, а время до проскока - временем защитного действия. Зависимость концентрации данного компонента за слоем от времени наз. выходной кривой. Эти кривые служат осн. эксперим. материалом, позволяющим судить о закономерностях динамики адсорбция
Аппаратурное оформление адсорбционных процессов
Существует множество технол. приемов проведения адсорбц. процессов. Широко распространены циклич. (перио-дич.) установки с неподвижным слоем адсорбента, осн. узел которых - один или неск. адсорберов, выполненных в виде полых колонн, заполняемых гранулированным адсорбентом. Газовый (или жидкостной) поток, содержащий адсорбируемые компоненты, пропускается через слой адсорбента до проскокадсорбция После этого адсорбент в адсорбере регенерируют, а газовый поток направляют в др. адсорбер. Регенерация адсорбента включает ряд стадий, из которых ос новная-десорбция, т.е. выделение ранее поглощенного в-ва из адсорбентадсорбция Десорбцию проводят нагреванием, сбросом давления в газовой фазе, вытеснением (напр., острым водяным паром) или комбинацией этих методов. Т. к. времена адсорбция и регенерации не совпадают, подбирают такое число одновременно работающих и регенерируемых адсорберов, чтобы в целом процесс шел непрерывно.
По техн. и экономич. соображениям регенерацию не доводят до концадсорбция Поэтому рабочая емкость адсорбента равна разности между максимально достигаемой в данных условиях адсорбция и кол-вом адсорбата, остающегося в адсорбенте после регенерации. Вследствие этого изотермы адсорбция , соответствующие процессу в адсорбере, не должны быть слишком крутыми.
В описанной схеме возможны два варианта: 1) целевой продукт адсорбируется из газового потока практически полностью, и тогда он содержится в десорбате, откуда его извлекают тем или иным способом; 2) целевой продукт адсорбируется хуже, чем др. компоненты газовой смеси, и тогда он содержится в выходящем газовом потоке. По первому варианту работают, например, рекуперационные установки на вискозных произ-вах, улавливающие из отходящих газов и возвращающие в цикл CS 2 . Производительность таких установок достигает сотен тысяч м 3 очищаемого газа в час; адсорбент-активный уголь с не слишком тонкими микропорами, т.е. уголь, в котором константа E по ТОЗМ (см. выше) равна 20-25 кДж/моль. Это значение E 0 соответствует не слишком крутой изотерме, что обеспечивает хорошие условия регенерации. Такие угли наз. рекуперационными. Десорбцию осуществляют острым водяным паром. Для экономии энергии холодные и газовые горячие потоки пропускают через теплообменники.
Очень важна осушка газов и жидкостей, например нефтяных газов перед их переработкой или прир. газов перед транспортировкой; адсорбенты-силикагель или цеолиты. Десорбцию осуществляют нагреванием. Т. к. десорбция цеолита связана с большими энергозатратами, применяют комбинированный адсорбент: осн. массу влаги поглощают легко регенерируемым силикагелем, а глубокую доосушку-цеолитом.
При тепловой регенерации полный цикл включает адсорбция , нагрев адсорбента, его десорбцию и охлаждение. Большое число стадий обусловливает низкую интенсивность и высокую энергоемкость процессадсорбция Поэтому широкое распространение получили т. наз. короткоцикловые установки, весь цикл в которых занимает неск. минут. В них газ подается в адсорбер под значит. давлением, которое затем сбрасывается, и происходит десорбция. Весь процесс идет почти изотермически (отклонение от изотермичности вызывается только выделением теплоты адсорбция и поглощением теплоты при десорбции). Стадии цикла: адсорбция , сброс давления, десорбция, подъем давления. Пример-установки с цеолитом для получения воздуха, обогащенного кислородом.
В установках сдвижущимся слоем адсорбента (в т. наз. гиперсорберах) последний под действием силы тяжести медленно опускается, выводится из ниж. части адсорбера и попадает в т. наз. эрлифт, представляющий собой вертикальную трубу, параллельную адсорбц. колонне. По этой трубе снизу вверх движется поток воздуха, который поднимает зерна адсорбента в верх. часть колонны. Перерабатываемый газовый поток поступает в среднюю часть адсорбера и движется вверх противотоком к адсорбенту. В верхней части колонны непрерывно происходит адсорбция , в нижней - регенерация адсорбента (см. также Адсорбционная очистка).
В установках с псевдоожиженным ("кипящим") слоем адсорбента газовый поток, поступающий в адсорбер снизу, приводит адсорбент во взвешенное состояние. При этом резко увеличивается эффективность массообмена между адсорбентом и газом и сокращается длительность адсорбция и десорбции. Такие установки имеют высокую производительность. Их широкому распространению препятствуют высокие требования, предъявляемые к мех. прочности зерен адсорбента (недостаточная прочность обусловливает значит. потери адсорбента вследствие его истирания и уноса из аппарата).
Осн. требования к адсорбентам: большая адсорбц. емкость, т.е. они должны представлять собой дисперсные тела с большой уд. поверхностью или с большим объемом пор; хим. природа поверхности должна обеспечивать эффективную адсорбция данных в-в в данных условиях; хим. и термич. стойкость, регенерируемость, доступность. наиб. распространение получили активные угли, ксерогели некоторых оксидов (силика-гели, алюмогели и др.), цеолиты; из непористых адсорбентов-техн. углерод (сажа) и высокодисперсный SiO 2 (аэросил, "белая сажа").
Области применения адсорбционной техники
На явлении адсорбция основаны мн. способы очистки воздуха от вредных примесей (см. Газов очистка), воды (см. Водоподготовка), а также сахарных сиропов при сахароварении, фруктовых соков и др. жидкостей в пищ. пром-сти, отработанных смазочных масел. Удаление влаги как вредной примеси из газов и жидкостей с помощью твердых адсорбентов-одна из важных отраслей адсорбц. техники (см. также Газов осушка).
На адсорбц. процессах основано тонкое разделение смесей веществ и выделение из сложных смесей определенных компонентов. Примеры-разделение изомеров алканов с целью получения нормальных углеводородов для произ-ва ПАВ, разделение нефтей при произ-ве моторных топлив. Для газовых смесей адсорбц. методы разделения используют при получении воздуха, обогащенного кислородом (вплоть до почти чистого О 2); во мн. случаях эти методы успешно конкурируют с ректификационным (см. Воздуха разделение).
Быстро развивающаяся область применения адсорбц. техники-медицина, где она служит для извлечения вредных веществ из крови (метод гемосорбции) и др. физиол. жидкостей. Высокие требования к стерильности ставят очень трудную задачу подбора подходящих адсорбентов. К ним относятся специально приготовленные активные угли.
Лит.: Брунауэр С., Адсорбция газов и паров, пер. с англ., т. 1, М., 1948; де Бур Я, Динамический характер адсорбции, пер. с англ., М., 1962; Адсорбция и пористость, под ред. М. М. Дубинина [и др.], М., 1976; Кельиев Н. В., Основы адсорбционной техники, 2 изд., М., 1984; Young D.M., Crowell A.D., Physical adsorption of gases, L., 1962. М.М.Дубинин, В.В. Серпинский.
Выберите первую букву в названии статьи:
Адсорбция имеет место на границе раздела фаз. Поэтому термодинамическое описание поверхностных явлений елесообразно рассматривать как частный случай термодинамики гетерогенных систем.
Рис. 3.4. Адсорбция по Гиббсу:1- двухфазная система сравнения, 2- реальная двухфазная система с неоднородной областью
В термодинамике гетерогенных систем используется принцип аддитивности , который заключается в следующем: все экстенсивные свойства гетерогенной системы равны сумме соответствующих экстенсивных свойств, которыми обладали бы фазы до того, как их привели в контакт. Обозначим фазы через α и β (рис.4). Тогда для идеальной системы, такой, что свойства фаз вблизи поверхности раздела совпадают с их объемными свойствами, для внутренней энергии U, объема V, массы (числа молей) n, энтропии S после установления равновесия в гетерогенной системе справедливы соотношения:
U = U α + U β , V = V α + V β , n = n α + n β , S = S α + S β
При этом подразумевается, что температура и давление в обеих фазах одинаковы.
Для реальных гетерогенных систем переходная область на границе двух фаз вносит дополнительный вклад в экстенсивные свойства системы. Если поверхностные явления имеют место, следует учитывать отличие экстенсивных свойств реальной гетерогенной системы от экстенсивных свойств модельной системы, в которой поверхностные явления отсутствуют. Такая система называется системой сравнения. Система сравнения обладает теми же интенсивными параметрами (T, P, C i …) и таким же объемом V, что и реальная система (рис. 4).
С термодинамической точки зрения под величиной адсорбции Г понимают избыточное количество вещества n s , выраженное в молях или граммах, которым обладает реальная гетерогенная система по сравнению с системой сравнения, отнесенное к площади поверхности раздела фаз или к площади поверхности адсорбента А. Принимается, что система сравнения обладает теми же интенсивными параметрами (T, P, C i), и таким же объемом (V = V α + V β), что и реальная система (рис.4).
Г = (n - n α - n β)/A = n s /A 3.11
Избыточные термодинамические функции переходной области реальной системы (обозначим их индексом s) можно записать как
U s = U - U α - U β , n s = n - n α - n β , S s = S - S α - S β и т.д.
Экспериментальные измерения адсорбции всегда дают адсорбцию именно как избыток компонента в реальной системе по сравнению с выбранной системой сравнения. Например, при адсорбции газа на твердом адсорбенте или при адсорбции компонентов на твердой фазе для нахождения величин адсорбции определяют изменение начальных концентраций адсорбата после соприкосновения фаз α и β
n i s = V(C i o - C i),
где C i o – исходные концентрация i –го компонента, C i – концентрация i – го компонента после установления равновесия между соприкасающимися фазами. При этом считается, что объем V не меняется. Однако, концентрация i -го компонента C i , полученная экспериментально, определяется в объеме V’ над поверхностью раздела фаз без учета объема неоднородной области переходного слоя V α у границы раздела, где концентрация составляет C i α . Таким образом, ввиду существования в реальной системе неоднородной области, общий объем системы можно представить как V = V’ + V α . Всё количество i –го компонента C i o распределится между этими двумя объемами:
V C i o = V’ C i + V α C i α ,
и число молей компонента i , адсорбированного на поверхности раздела фаз, будет равно
n i s = (V’C i + V α C i α) – (V’ + V α)C i = V α (C i α – C i) 3.12
Т.е. определяемая экспериментально адсорбция есть избыток i-го компонента в объеме V α по сравнению с количеством этого компонента в таком же объеме вдали от поверхности раздела фаз. Именно такая адсорбция называется адсорбцией по Гиббсу .
V α C i α называется полным содержанием i- го компонента в адсорбционном слое. В области очень малых концентрацийC i в объеме V’ поправкой V α C i уравнения (3.2) можно пренебречь и считать измеренную величину V α C i α полным содержанием i- го компонента в адсорбционном слое, например, при адсорбции газа на твердом адсорбенте при низких давлениях.


